3. Adaptive MPI (AMPI)¶
Contents
- Adaptive MPI (AMPI)
- Introduction
- Charm++
- AMPI
- AMPI Compliance to MPI Standards
- AMPI Extensions to MPI Standards
- Name for Main Program
- Global Variable Privatization
- Extensions for Migrations
- Extensions for Checkpointing
- Extensions for Memory Efficiency
- Extensions for Interoperability
- Charm++ Interoperability
- Extensions for Sequential Re-run of a Parallel Node
- User Defined Initial Mapping
- Performance Visualization
- Compiling AMPI Programs
- AMPI Example Applications
- Installing AMPI
- Building and Running AMPI Programs
3.1. Introduction¶
This manual describes Adaptive MPI (AMPI), which is an implementation of the MPI standard [1] on top of Charm++. AMPI acts as a regular MPI implementation (akin to MPICH, OpenMPI, MVAPICH, etc.) with several built-in extensions that allow MPI developers to take advantage of Charm++’s dynamic runtime system, which provides support for process virtualization, overlap of communication and computation, load balancing, and fault tolerance with zero to minimal changes to existing MPI codes.
In this manual, we first describe the philosophy behind Adaptive MPI, then give a brief introduction to Charm++ and rationale for AMPI. We then describe AMPI in detail. Finally we summarize the changes required for existing MPI codes to run with AMPI. Appendices contain the details of installing AMPI, and building and running AMPI programs.
3.1.1. Overview¶
Developing parallel Computational Science and Engineering (CSE) applications is a complex task. One has to implement the right physics, develop or choose and code appropriate numerical methods, decide and implement the proper input and output data formats, perform visualizations, and be concerned with correctness and efficiency of the programs. It becomes even more complex for multi-physics coupled simulations, many of which are dynamic and adaptively refined so that load imbalance becomes a major challenge. In addition to imbalance caused by dynamic program behavior, hardware factors such as latencies, variability, and failures must be tolerated by applications. Our philosophy is to lessen the burden of application developers by providing advanced programming paradigms and versatile runtime systems that can handle many common programming and performance concerns automatically and let application programmers focus on the actual application content.
Many of these concerns can be addressed using the processor virtualization and over-decomposition philosophy of Charm++. Thus, the developer only sees virtual processors and lets the runtime system deal with underlying physical processors. This is implemented in AMPI by mapping MPI ranks to Charm++ user-level threads as illustrated in Figure 3. As an immediate and simple benefit, the programmer can use as many virtual processors (“MPI ranks”) as the problem can be easily decomposed to. For example, suppose the problem domain has \(n*2^n\) parts that can be easily distributed but programming for general number of MPI processes is burdensome, then the developer can have \(n*2^n\) virtual processors on any number of physical ones using AMPI.
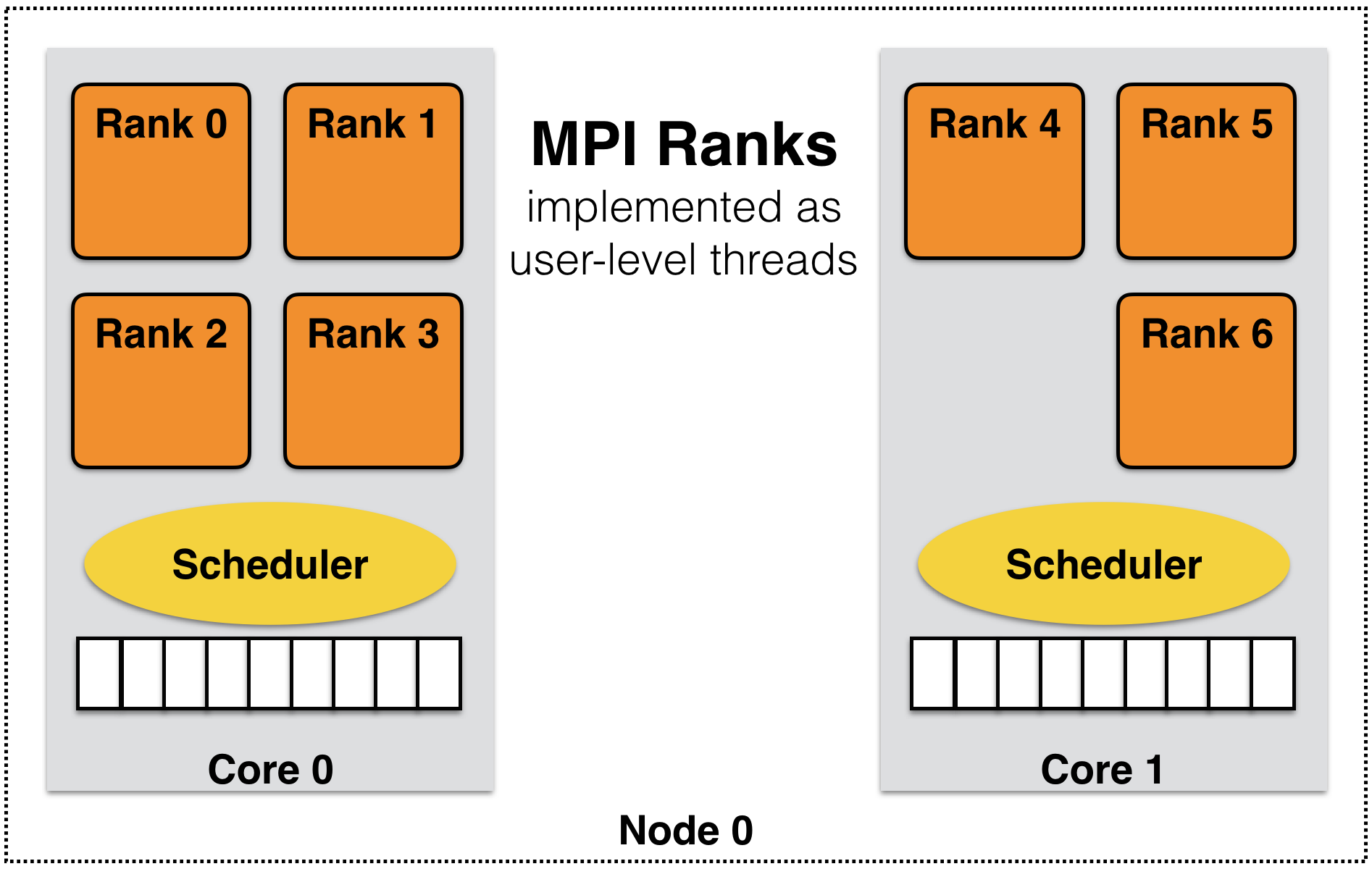
3 MPI ranks are implemented as user-level threads in AMPI rather than Operating System processes.
AMPI’s execution model consists of multiple user-level threads per Processing Element (PE). The Charm++ scheduler coordinates execution of these user-level threads (also called Virtual Processors or VPs) and controls execution. These VPs can also migrate between PEs for the purpose of load balancing or other reasons. The number of VPs per PE specifies the virtualization ratio (degree of over-decomposition). For example, in Figure 3 the virtualization ratio is \(3.5\) (there are four VPs on PE 0 and three VPs on PE 1). Figure 4 shows how the problem domain can be over-decomposed in AMPI’s VPs as opposed to other MPI implementations.

4 The problem domain is over-decomposed to more VPs than PEs.
Another benefit of virtualization is communication and computation overlap, which is automatically realized in AMPI without programming effort. Techniques such as software pipelining require significant programming effort to achieve this goal and improve performance. However, one can use AMPI to have more virtual processors than physical processors to overlap communication and computation. Each time a VP is blocked for communication, the Charm++ scheduler picks the next VP among those that are ready to execute. In this manner, while some of the VPs of a physical processor are waiting for a message to arrive, others can continue their execution. Thus, performance improves without any changes to the application source code.
Another potential benefit is that of better cache utilization. With over-decomposition, a smaller subdomain is accessed by a VP repeatedly in different function calls before getting blocked by communication and switching to another VP. That smaller subdomain may fit into cache if over-decomposition is enough. This concept is illustrated in Figure 3 where each AMPI rank’s subdomain is smaller than the corresponding MPI subdomain and so may fit into cache memory. Thus, there is a potential performance improvement without changing the source code.
One important concern is that of load imbalance. New generation parallel applications are dynamically varying, meaning that processors’ load is shifting during execution. In a dynamic simulation application such as rocket simulation, burning solid fuel, sub-scaling for a certain part of the mesh, crack propagation, particle flows all contribute to load imbalance. A centralized load balancing strategy built into an application is impractical since each individual module is developed mostly independently by various developers. In addition, embedding a load balancing strategy in the code complicates it greatly, and programming effort increases significantly. The runtime system is uniquely positioned to deal with load imbalance. Figure 5 shows the runtime system migrating a VP after detecting load imbalance. This domain may correspond to a weather forecast model where there is a storm cell in the top-left quadrant, which requires more computation to simulate. AMPI will then migrate VP 1 to balance the division of work across processors and improve performance. Note that incorporating this sort of load balancing inside the application code may take a lot of effort and complicate the code.
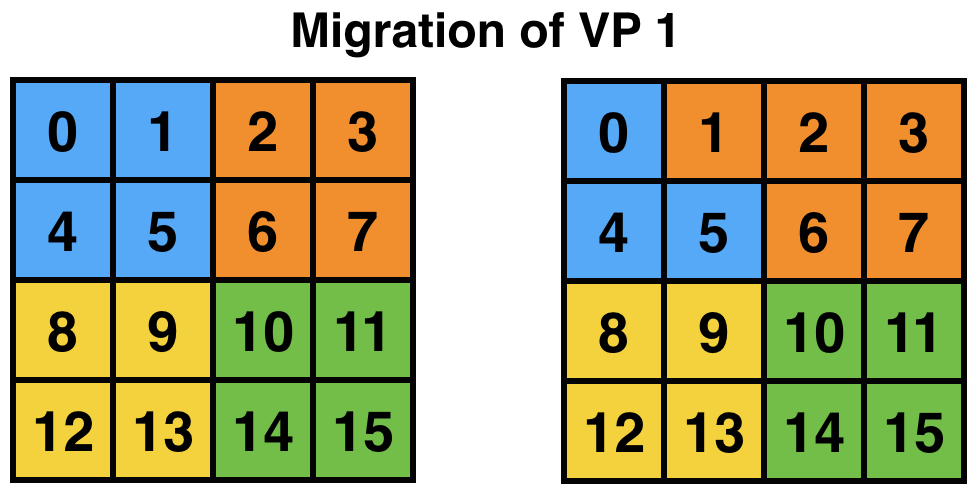
5 AMPI can migrate VPs across processes for load balancing.
There are many different load balancing strategies built into Charm++ that can be selected by an AMPI application developer. Among those, some may fit better for a particular application depending on its characteristics. Moreover, one can write a new load balancer, best suited for an application, by the simple API provided inside Charm++ infrastructure. Our approach is based on actual measurement of load information at runtime, and on migrating computations from heavily loaded to lightly loaded processors.
For this approach to be effective, we need the computation to be split into pieces many more in number than available processors. This allows us to flexibly map and re-map these computational pieces to available processors. This approach is usually called “multi-domain decomposition”.
Charm++, which we use as a runtime system layer for the work described here, simplifies our approach. It embeds an elaborate performance tracing mechanism, a suite of plug-in load balancing strategies, infrastructure for defining and migrating computational load, and is interoperable with other programming paradigms.
3.2. Charm++¶
Charm++ is an object-oriented parallel programming library for C. It differs from traditional message passing programming libraries (such as MPI) in that Charm++ is “message-driven”. Message-driven parallel programs do not block the processor waiting for a message to be received. Instead, each message carries with itself a computation that the processor performs on arrival of that message. The underlying runtime system of Charm++ is called Converse, which implements a “scheduler” that chooses which message to schedule next (message-scheduling in Charm++ involves locating the object for which the message is intended, and executing the computation specified in the incoming message on that object). A parallel object in Charm++ is a C object on which a certain computations can be asked to be performed from remote processors.
Charm++ programs exhibit latency tolerance since the scheduler always picks up the next available message rather than waiting for a particular message to arrive. They also tend to be modular, because of their object-based nature. Most importantly, Charm++ programs can be dynamically load balanced, because the messages are directed at objects and not at processors; thus allowing the runtime system to migrate the objects from heavily loaded processors to lightly loaded processors.
Since many CSE applications are originally written using MPI, one would have to rewrite existing code if they were to be converted to Charm++ to take advantage of dynamic load balancing and other Charm++ features. This is indeed impractical. However, Converse - the runtime system of Charm++ - supports interoperability between different parallel programming paradigms such as parallel objects and threads. Using this feature, we developed AMPI, which is described in more detail in the next section.
3.3. AMPI¶
AMPI utilizes the dynamic load balancing and other capabilities of Charm++ by associating a “user-level” thread with each Charm++ migratable object. User’s code runs inside this thread, so that it can issue blocking receive calls similar to MPI, and still present the underlying scheduler an opportunity to schedule other computations on the same processor. The runtime system keeps track of the computational loads of each thread as well as the communication graph between AMPI threads, and can migrate these threads in order to balance the overall load while simultaneously minimizing communication overhead.
3.3.1. AMPI Compliance to MPI Standards¶
Currently AMPI supports the MPI-2.2 standard, with preliminary support for most MPI-3.1 features and a collection of extensions explained in detail in this manual. One-sided communication calls in MPI-2 and MPI-3 are implemented, but they do not yet take advantage of RMA features. Non-blocking collectives have been defined in AMPI since before MPI-3.0’s adoption of them. Also ROMIO [2] has been integrated into AMPI to support parallel I/O features.
3.3.2. AMPI Extensions to MPI Standards¶
The following are AMPI extensions to the MPI standard, which will be
explained in detail in this manual. All AMPI extensions to the MPI
standard are prefixed with AMPI_ rather than MPI_. All
extensions are available in C, C++, and Fortran, with the exception of
AMPI_Command_argument_count and AMPI_Get_command_argument which
are only available in Fortran.
AMPI_Migrate AMPI_Register_pup AMPI_Get_pup_data
AMPI_Migrate_to_pe AMPI_Set_migratable AMPI_Evacuate
AMPI_Load_set_value AMPI_Load_start_measure AMPI_Load_stop_measure
AMPI_Iget AMPI_Iget_wait AMPI_Iget_data
AMPI_Iget_free AMPI_Type_is_contiguous AMPI_Register_main
AMPI_Yield AMPI_Suspend AMPI_Resume
AMPI_Alltoall_medium AMPI_Alltoall_long
AMPI_Register_just_migrated AMPI_Register_about_to_migrate
AMPI_Command_argument_count AMPI_Get_command_argument
AMPI provides a set of built-in attributes on all communicators and
windows to find the number of the worker thread, process, or host that a
rank is currently running on, as well as the total number of worker
threads, processes, and hosts in the job. We define a worker thread to
be a thread on which one of more AMPI ranks are scheduled. We define a
process here as an operating system process, which may contain one or
more worker threads. The built-in attributes are AMPI_MY_WTH,
AMPI_MY_PROCESS, AMPI_NUM_WTHS, and AMPI_NUM_PROCESSES.
These attributes are accessible from any rank by calling
MPI_Comm_get_attr, such as:
! Fortran:
integer :: my_wth, flag, ierr
call MPI_Comm_get_attr(MPI_COMM_WORLD, AMPI_MY_WTH, my_wth, flag, ierr)
// C/C++:
int my_wth, flag;
MPI_Comm_get_attr(MPI_COMM_WORLD, AMPI_MY_WTH, &my_wth, &flag);
AMPI also provides extra communicator types that users can pass to
MPI_Comm_split_type: AMPI_COMM_TYPE_HOST for splitting a
communicator into disjoint sets of ranks that share the same physical
host, AMPI_COMM_TYPE_PROCESS for splitting a communicator into
disjoint sets of ranks that share the same operating system process, and
AMPI_COMM_TYPE_WTH, for splitting a communicator into disjoint sets
of ranks that share the same worker thread.
For parsing Fortran command line arguments, AMPI Fortran programs should use our extension APIs, which are similar to Fortran 2003’s standard APIs. For example:
integer :: i, argc, ierr
integer, parameter :: arg_len = 128
character(len=arg_len), dimension(:), allocatable :: raw_arguments
call AMPI_Command_argument_count(argc)
allocate(raw_arguments(argc))
do i = 1, size(raw_arguments)
call AMPI_Get_command_argument(i, raw_arguments(i), arg_len, ierr)
end do
3.3.3. Name for Main Program¶
To convert an existing program to use AMPI, the main function or program may need to be renamed. The changes should be made as follows:
3.3.3.1. Fortran¶
You must declare the main program as a subroutine called “MPI_MAIN”. Do not declare the main subroutine as a program because it will never be called by the AMPI runtime.
program pgm -> subroutine MPI_Main
... ...
end program -> end subroutine
3.3.3.2. C or C++¶
The main function can be left as is, if mpi.h is included before the
main function. This header file has a preprocessor macro that renames
main, and the renamed version is called by the AMPI runtime by each
thread.
3.3.4. Global Variable Privatization¶
For the before-mentioned benefits to be effective, one needs to map multiple user-level threads onto each processor. Traditional MPI programs assume that the entire processor is allocated to themselves, and that only one thread of control exists within the process’s address space. So, they may safely use global and static variables in the program. However, global and static variables are problematic for multi-threaded environments such as AMPI or OpenMP. This is because there is a single instance of those variables so they will be shared among different threads in the single address space, so if programmers are not careful a wrong result may be produced by the program. Figure 6 shows an example of a multi-threaded application with two threads in a single process. \(var\) is a global or static variable in this example. Thread 1 assigns a value to it, then it gets blocked for communication and another thread can continue. Thereby, thread 2 is scheduled next and accesses \(var\) which is wrong. The semantics of this program needs separate instances of \(var\) for each of the threads. That is where the need arises to make some transformations to the original MPI program in order to run correctly with AMPI. Note, this is the only change necessary to run an MPI program with AMPI, that the program be thread-safe and have no global or static variables whose values differ across different MPI ranks. Also note that global variables that are constant or are only written to once to the same value across all ranks during initialization are already thread-safe.

6 Mutable global or static variables are an issue for AMPI
The basic transformation needed to port the MPI program to AMPI is privatization of global variables. With the MPI process model, each MPI node can keep a copy of its own “permanent variables” - variables that are accessible from more than one subroutines without passing them as arguments. Module variables, “saved” subroutine local variables, and common blocks in Fortran90 belong to this category. If such a program is executed without privatization on AMPI, all the AMPI threads that reside in the same process will access the same copy of such variables, which is clearly not the desired semantics. To ensure correct execution of the original source program, it is necessary to make such variables “private” to individual threads. We provide three choices with varying degrees of developer effort required and varying degrees of portability: manual encapsulation of global state, a thread-local storage based automated mechanism, and global offset table based automated mechanism.
3.3.4.1. Automatic Thread-Local Storage Swapping¶
Thread Local Store (TLS) was originally employed in kernel threads to localize variables to threads and provide thread safety. It can be used by annotating global/static variable declarations in C with thread_local, in C with __thread or C11 with thread_local or _Thread_local, and in Fortran with OpenMP’s threadprivate attribute. OpenMP is required for using tlsglobals in Fortran code since Fortran has no other method of using TLS. The __thread keyword is not an official extension of the C language, though compiler writers are encouraged to implement this feature.
It handles both global and static variables and has no context-switching overhead. AMPI provides runtime support for privatizing thread-local variables to user-level threads by changing the TLS segment register when context switching between user-level threads. The runtime overhead is that of changing a single pointer per user-level thread context switch. Currently, Charm++ supports it for x86/x86_64 platforms when using GNU compilers.
// C/C++ example:
int myrank;
double xyz[100];
! Fortran example:
integer :: myrank
real*8, dimension(100) :: xyz
For the example above, the following changes to the code handle the global variables:
// C++ example:
thread_local int myrank;
thread_local double xyz[100];
// C example:
__thread int myrank;
__thread double xyz[100];
! Fortran example:
integer :: myrank
real*8, dimension(100) :: xyz
!$omp threadprivate(myrank)
!$omp threadprivate(xyz)
The runtime system also should know that TLS-Globals is used at both compile and link time:
$ ampicxx -o example example.C -tlsglobals
3.3.4.2. Automatic Process-in-Process Runtime Linking Privatization¶
Process-in-Process (PiP) [PiP2018] Globals allows fully automatic privatization of global variables on GNU/Linux systems without modification of user code. All languages (C, C++, Fortran, etc.) are supported. This method currently lacks support for checkpointing and migration, which are necessary for load balancing and fault tolerance. Additionally, overdecomposition is limited to approximately 12 virtual ranks per logical node, though this can be resolved by building a patched version of glibc.
This method works by combining a specific method of building binaries
with a GNU extension to the dynamic linker. First, AMPI’s toolchain
wrapper compiles your user program as a Position Independent Executable
(PIE) and links it against a special shim of function pointers instead
of the normal AMPI runtime. It then builds a small loader utility that
links directly against AMPI. For each rank, this loader calls the
glibc-specific function dlmopen on the PIE binary with a unique
namespace index. The loader uses dlsym to populate the PIE binary’s
function pointers and then it calls the entry point. This dlmopen
and dlsym process repeats for each rank. As soon as execution jumps
into the PIE binary, any global variables referenced within will appear
privatized. This is because PIE binaries locate the global data segment
immediately after the code segment so that PIE global variables are
accessed relative to the instruction pointer, and because dlmopen
creates a separate copy of these segments in memory for each unique
namespace index.
Optionally, the first step in using PiP-Globals is to build PiP-glibc to
overcome the limitation on rank count per process. Use the instructions
at https://github.com/RIKEN-SysSoft/PiP/blob/pip-1/INSTALL to download
an installable PiP package or build PiP-glibc from source by following
the Patched GLIBC section. AMPI may be able to automatically detect
PiP’s location if installed as a package, but otherwise set and export
the environment variable PIP_GLIBC_INSTALL_DIR to the value of
<GLIBC_INSTALL_DIR> as used in the above instructions. For example:
$ export PIP_GLIBC_INSTALL_DIR=~/pip
To use PiP-Globals in your AMPI program (with or without PiP-glibc),
compile and link with the -pipglobals parameter:
$ ampicxx -o example.o -c example.cpp -pipglobals
$ ampicxx -o example example.o -pipglobals
No further effort is needed. Global variables in example.cpp will be
automatically privatized when the program is run. Any libraries and
shared objects compiled as PIE will also be privatized. However, if
these objects call MPI functions, it will be necessary to build them
with the AMPI toolchain wrappers, -pipglobals, and potentially also
the -standalone parameter in the case of shared objects. It is
recommended to do this in any case so that AMPI can ensure everything is
built as PIE.
Potential future support for checkpointing and migration will require
modification of the ld-linux.so runtime loader to intercept mmap
allocations of the previously mentioned segments and redirect them
through Isomalloc. The present lack of support for these features mean
PiP-Globals is best suited for testing AMPI during exploratory phases
of development, and for production jobs not requiring load balancing or
fault tolerance.
3.3.4.3. Automatic Filesystem-Based Runtime Linking Privatization¶
Filesystem Globals (FS-Globals) was discovered during the development of PiP-Globals and the two are highly similar. Like PiP-Globals, it requires no modification of user code and works with any language. It also currently lacks support for checkpointing and migration, preventing use of load balancing and fault tolerance. Unlike PiP-Globals, it is portable beyond GNU/Linux and has no limits to overdecomposition beyond available disk space.
FS-Globals works in the same way as PiP-Globals except that instead of
specifying namespaces using dlmopen, which is a GNU/Linux-specific
feature, this method creates copies of the user’s PIE binary on the
filesystem for each rank and calls the POSIX-standard dlopen.
To use FS-Globals, compile and link with the -fsglobals parameter:
$ ampicxx -o example.o -c example.cpp -fsglobals
$ ampicxx -o example example.o -fsglobals
No additional steps are required. Global variables in example.cpp
will be automatically privatized when the program is run. Variables in
statically linked libraries will also be privatized if compiled as PIE.
It is recommended to achieve this by building with the AMPI toolchain
wrappers and -fsglobals, and this is necessary if the libraries call
MPI functions. Shared objects are currently not supported by FS-Globals
due to the extra overhead of iterating through all dependencies and
copying each one per rank while avoiding system components, plus the
complexity of ensuring each rank’s program binary sees the proper set of
objects.
This method’s use of the filesystem is a drawback in that it is slow during startup and can be considered wasteful. Additionally, support for load balancing and fault tolerance would require further development in the future, using the same infrastructure as what PiP-Globals would require. For these reasons FS-Globals is best suited for the R&D phase of AMPI program development and for small jobs, and it may be less suitable for large production environments.
3.3.4.4. Automatic Global Offset Table Swapping¶
Thanks to the ELF Object Format, we have successfully automated the procedure of switching the set of user global variables when switching thread contexts. Executable and Linkable Format (ELF) is a common standard file format for Object Files in Unix-like operating systems. ELF maintains a Global Offset Table (GOT) for globals so it is possible to switch GOT contents at thread context-switch by the runtime system.
The only thing that the user needs to do is pass the flag
-swapglobals at both compile and link time (e.g. “ampicc -o prog
prog.c -swapglobals”). This method does not require any changes to the
source code and works with any language (C, C++, Fortran, etc). However,
it does not handle static variables, has a context switching overhead
that grows with the number of global variables, and is incompatible with
SMP builds of AMPI, where multiple virtual ranks can execute
simultaneously on different scheduler threads within an OS process.
Currently, this feature only works on x86 and x86_64 platforms that
fully support ELF, and it requires ld version 2.23 or older, or else a
patched version of ld 2.24+ that we provide here:
https://charm.cs.illinois.edu/gerrit/gitweb?p=libbfd-patches.git;a=tree;f=swapglobals
3.3.4.5. Manual Change¶
We have employed a strategy of argument passing to do this privatization transformation. That is, the global variables are bunched together in a single user-defined type, which is allocated by each thread dynamically or on the stack. Then a pointer to this type is passed from subroutine to subroutine as an argument. Since the subroutine arguments are passed on the stack, which is not shared across all threads, each subroutine when executing within a thread operates on a private copy of the global variables.
This scheme is demonstrated in the following examples. The original
Fortran90 code contains a module shareddata. This module is used in
the MPI_MAIN subroutine and a subroutine subA. Note that
PROGRAM PGM was renamed to SUBROUTINE MPI_MAIN and END PROGRAM
was renamed to END SUBROUTINE.
!FORTRAN EXAMPLE
MODULE shareddata
INTEGER :: myrank
DOUBLE PRECISION :: xyz(100)
END MODULE
SUBROUTINE MPI_MAIN ! Previously PROGRAM PGM
USE shareddata
include 'mpif.h'
INTEGER :: i, ierr
CALL MPI_Init(ierr)
CALL MPI_Comm_rank(MPI_COMM_WORLD, myrank, ierr)
DO i = 1, 100
xyz(i) = i + myrank
END DO
CALL subA
CALL MPI_Finalize(ierr)
END SUBROUTINE ! Previously END PROGRAM
SUBROUTINE subA
USE shareddata
INTEGER :: i
DO i = 1, 100
xyz(i) = xyz(i) + 1.0
END DO
END SUBROUTINE
//C Example
#include <mpi.h>
int myrank;
double xyz[100];
void subA();
int main(int argc, char** argv){
int i;
MPI_Init(&argc, &argv);
MPI_Comm_rank(MPI_COMM_WORLD, &myrank);
for(i=0;i<100;i++)
xyz[i] = i + myrank;
subA();
MPI_Finalize();
}
void subA(){
int i;
for(i=0;i<100;i++)
xyz[i] = xyz[i] + 1.0;
}
AMPI executes the main subroutine inside a user-level thread as a subroutine.
Now we transform this program using the argument passing strategy. We first group the shared data into a user-defined type.
!FORTRAN EXAMPLE
MODULE shareddata
TYPE chunk ! modified
INTEGER :: myrank
DOUBLE PRECISION :: xyz(100)
END TYPE ! modified
END MODULE
//C Example
struct shareddata{
int myrank;
double xyz[100];
};
Now we modify the main subroutine to dynamically allocate this data and
change the references to them. Subroutine subA is then modified to
take this data as argument.
!FORTRAN EXAMPLE
SUBROUTINE MPI_Main
USE shareddata
USE AMPI
INTEGER :: i, ierr
TYPE(chunk), pointer :: c ! modified
CALL MPI_Init(ierr)
ALLOCATE(c) ! modified
CALL MPI_Comm_rank(MPI_COMM_WORLD, c%myrank, ierr)
DO i = 1, 100
c%xyz(i) = i + c%myrank ! modified
END DO
CALL subA(c)
CALL MPI_Finalize(ierr)
END SUBROUTINE
SUBROUTINE subA(c)
USE shareddata
TYPE(chunk) :: c ! modified
INTEGER :: i
DO i = 1, 100
c%xyz(i) = c%xyz(i) + 1.0 ! modified
END DO
END SUBROUTINE
//C Example
void MPI_Main{
int i,ierr;
struct shareddata *c;
ierr = MPI_Init();
c = (struct shareddata*)malloc(sizeof(struct shareddata));
ierr = MPI_Comm_rank(MPI_COMM_WORLD, c.myrank);
for(i=0;i<100;i++)
c.xyz[i] = i + c.myrank;
subA(c);
ierr = MPI_Finalize();
}
void subA(struct shareddata *c){
int i;
for(i=0;i<100;i++)
c.xyz[i] = c.xyz[i] + 1.0;
}
With these changes, the above program can be made thread-safe. Note that
it is not really necessary to dynamically allocate chunk. One could
have declared it as a local variable in subroutine MPI_Main. (Or for
a small example such as this, one could have just removed the
shareddata module, and instead declared both variables xyz and
myrank as local variables). This is indeed a good idea if shared
data are small in size. For large shared data, it would be better to do
heap allocation because in AMPI, the stack sizes are fixed at the
beginning (and can be specified from the command line) and stacks do not
grow dynamically.
3.3.4.6. Source-to-source Transformation¶
Another approach is to do the changes described in the previous scheme automatically. It means that we can use a tool to transform the source code to move global or static variables in an object and pass them around. This approach is portable across systems and compilers and may also improve locality and hence cache utilization. It also does not have the context-switch overhead of swapping globals. We have multiple tools for automating these transformations for different languages. Currently, there is a tool called Photran [3] for refactoring Fortran codes that can do this transformation. It is Eclipse-based and works by constructing Abstract Syntax Trees (ASTs) of the program. We also have a tool built on top of the ROSE compiler [4] that works for C/C++ and Fortran programs that is available upon request. It emits patches for all files containing global variables which can then be applied to the source code.
Table 3 shows portability of different schemes.
| Privatization Scheme | x86 | x86_64 | Mac OS | BG/Q | Windows | PPC | ARM7 |
|---|---|---|---|---|---|---|---|
| Transformation | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| GOT-Globals | Yes | Yes | No | No | No | Yes | Yes |
| TLS-Globals | Yes | Yes | Yes | No | Maybe | Maybe | Maybe |
| PiP-Globals | Yes | Yes | No | No | No | Yes | Yes |
| FS-Globals | Yes | Yes | Yes | No | Maybe | Yes | Yes |
3.3.5. Extensions for Migrations¶
AMPI provides fully automated support for migrating MPI ranks between nodes of a system without any application-specific code at all. We do so using a memory allocator, Isomalloc, that allocates memory per user-level thread to globally unique virtual memory addresses. This means that every worker thread in the system reserves slices of virtual memory for all user-level threads, allowing transparent migration of stacks and pointers into memory (Isomalloc requires 64-bit virtual memory addresses and support from the operating system for mapping memory to arbitrary virtual addresses). Applications only need to link with Isomalloc to enable automatic migratability, using -memory isomalloc.
For systems that do not support Isomalloc and for users that wish to have more fine-grain control over which application data structures will be copied at migration time, we have added a few calls to AMPI. These include the ability to register thread-specific data with the run-time system, to pack and unpack all of the thread’s data, and to express willingness to migrate.
3.3.5.1. Registering User Data¶
When the AMPI runtime system decides that load imbalance exists within
the application, it will invoke one of its internal load balancing
strategies, which determines the new mapping of AMPI ranks so as to
balance the load. Then the AMPI runtime packs up the rank’s state and
moves it to its new home processor. AMPI packs up any internal data in
use by the rank, including the thread’s stack in use. This means that
the local variables declared in subroutines in a rank, which are created
on stack, are automatically packed up by the AMPI runtime system.
However, it has no way of knowing what other data are in use by the
rank. Thus upon starting execution, a rank needs to notify the system
about the data that it is going to use (apart from local variables).
Even with the data registration, AMPI cannot determine what size the
data is, or whether the registered data contains pointers to other
places in memory. For this purpose, a packing subroutine also needs to
be provided to the AMPI runtime system along with registered data. (See
next section for writing packing subroutines.) The call provided by AMPI
for doing this is AMPI_Register_pup. This function takes three
arguments: a data item to be transported along with the rank, the pack
subroutine, and a pointer to an integer which denotes the registration
identifier. In C/C++ programs, it may be necessary to use this integer
value after migration completes and control returns to the rank with the
function AMPI_Get_pup_data.
3.3.5.2. Migration¶
The AMPI runtime system could detect load imbalance by itself and invoke the load balancing strategy. However, since the application code is going to pack/unpack the rank’s data, writing the pack subroutine will be complicated if migrations occur at a stage unknown to the application. For example, if the system decides to migrate a rank while it is in initialization stage (say, reading input files), application code will have to keep track of how much data it has read, what files are open etc. Typically, since initialization occurs only once in the beginning, load imbalance at that stage would not matter much. Therefore, we want the demand to perform load balance check to be initiated by the application.
AMPI provides a subroutine AMPI_Migrate(MPI_Info hints); for this
purpose. Each rank periodically calls AMPI_Migrate. Typical CSE
applications are iterative and perform multiple time-steps. One should
call AMPI_Migrate in each rank at the end of some fixed number of
timesteps. The frequency of AMPI_Migrate should be determined by a
tradeoff between conflicting factors such as the load balancing
overhead, and performance degradation caused by load imbalance. In some
other applications, where application suspects that load imbalance may
have occurred, as in the case of adaptive mesh refinement; it would be
more effective if it performs a couple of timesteps before telling the
system to re-map ranks. This will give the AMPI runtime system some time
to collect the new load and communication statistics upon which it bases
its migration decisions. Note that AMPI_Migrate does NOT tell the
system to migrate the rank, but merely tells the system to check the
load balance after all the ranks call AMPI_Migrate. To migrate the
rank or not is decided only by the system’s load balancing strategy.
Essentially, a call to AMPI_Migrate signifies to the runtime system
that the application has reached a point at which it is safe to
serialize the local state. Knowing this, the runtime system can act in
several ways.
The MPI_Info object taken as a parameter by AMPI_Migrate gives users
a way to influence the runtime system’s decision-making and behavior.
AMPI provides two built-in MPI_Info objects for this, called
AMPI_INFO_LB_SYNC and AMPI_INFO_LB_ASYNC. Synchronous load
balancing assumes that the application is already at a synchronization
point. Asynchronous load balancing does not assume this.
Calling AMPI_Migrate on a rank with pending send requests (i.e. from
MPI_Isend) is currently not supported, therefore users should always
wait on any outstanding send requests before calling AMPI_Migrate.
// Main time-stepping loop
for (int iter=0; iter < max_iters; iter++) {
// Time step work ...
if (iter % lb_freq == 0)
AMPI_Migrate(AMPI_INFO_LB_SYNC);
}
Note that migrating ranks around the cores and nodes of a system can
change which ranks share physical resources, such as memory. A
consequence of this is that communicators created via
MPI_Comm_split_type are invalidated by calls to AMPI_Migrate
that result in migration which breaks the semantics of that communicator
type. The only valid routine to call on such communicators is
MPI_Comm_free.
We also provide callbacks that user code can register with the runtime
system to be invoked just before and right after migration:
AMPI_Register_about_to_migrate and AMPI_Register_just_migrated
respectively. Note that the callbacks are only invoked on those ranks
that are about to actually migrate or have just actually migrated.
AMPI provide routines for starting and stopping load measurements, and
for users to explicitly set the load value of a rank using the
following: AMPI_Load_start_measure, AMPI_Load_stop_measure,
AMPI_Load_reset_measure, and AMPI_Load_set_value. And since AMPI
builds on top of Charm++, users can experiment with the suite of load
balancing strategies included with Charm++, as well as write their own
strategies based on user-level information and heuristics.
3.3.5.3. Packing/Unpacking Thread Data¶
Once the AMPI runtime system decides which ranks to send to which
processors, it calls the specified pack subroutine for that rank, with
the rank-specific data that was registered with the system using
AMPI_Register_pup. If an AMPI application uses Isomalloc, then the
system will define the Pack/Unpack routines for the user. This section
explains how a subroutine should be written for performing explicit
pack/unpack.
There are three steps for transporting the rank’s data to another processor. First, the system calls a subroutine to get the size of the buffer required to pack the rank’s data. This is called the “sizing” step. In the next step, which is called immediately afterward on the source processor, the system allocates the required buffer and calls the subroutine to pack the rank’s data into that buffer. This is called the “packing” step. This packed data is then sent as a message to the destination processor, where first a rank is created (along with the thread) and a subroutine is called to unpack the rank’s data from the buffer. This is called the “unpacking” step.
Though the above description mentions three subroutines called by the AMPI runtime system, it is possible to actually write a single subroutine that will perform all the three tasks. This is achieved using something we call a “pupper”. A pupper is an external subroutine that is passed to the rank’s pack-unpack-sizing subroutine, and this subroutine, when called in different phases performs different tasks. An example will make this clear:
Suppose the user data, chunk, is defined as a derived type in Fortran90:
!FORTRAN EXAMPLE
MODULE chunkmod
INTEGER, parameter :: nx=4, ny=4, tchunks=16
TYPE, PUBLIC :: chunk
REAL(KIND=8) t(22,22)
INTEGER xidx, yidx
REAL(KIND=8), dimension(400):: bxm, bxp, bym, byp
END TYPE chunk
END MODULE
//C Example
struct chunk{
double t;
int xidx, yidx;
double bxm,bxp,bym,byp;
};
Then the pack-unpack subroutine chunkpup for this chunk module is
written as:
!FORTRAN EXAMPLE
SUBROUTINE chunkpup(p, c)
USE pupmod
USE chunkmod
IMPLICIT NONE
INTEGER :: p
TYPE(chunk) :: c
call pup(p, c%t)
call pup(p, c%xidx)
call pup(p, c%yidx)
call pup(p, c%bxm)
call pup(p, c%bxp)
call pup(p, c%bym)
call pup(p, c%byp)
end subroutine
//C Example
void chunkpup(pup_er p, struct chunk c){
pup_double(p,c.t);
pup_int(p,c.xidx);
pup_int(p,c.yidx);
pup_double(p,c.bxm);
pup_double(p,c.bxp);
pup_double(p,c.bym);
pup_double(p,c.byp);
}
There are several things to note in this example. First, the same
subroutine pup (declared in module pupmod) is called to
size/pack/unpack any type of data. This is possible because of procedure
overloading possible in Fortran90. Second is the integer argument p.
It is this argument that specifies whether this invocation of subroutine
chunkpup is sizing, packing or unpacking. Third, the integer
parameters declared in the type chunk need not be packed or unpacked
since they are guaranteed to be constants and thus available on any
processor.
A few other functions are provided in module pupmod. These functions
provide more control over the packing/unpacking process. Suppose one
modifies the chunk type to include allocatable data or pointers that
are allocated dynamically at runtime. In this case, when chunk is
packed, these allocated data structures should be deallocated after
copying them to buffers, and when chunk is unpacked, these data
structures should be allocated before copying them from the buffers. For
this purpose, one needs to know whether the invocation of chunkpup
is a packing one or unpacking one. For this purpose, the pupmod
module provides functions fpup_isdeleting(fpup_isunpacking).
These functions return logical value .TRUE. if the invocation is for
packing (unpacking), and .FALSE. otherwise. The following example
demonstrates this:
Suppose the type dchunk is declared as:
!FORTRAN EXAMPLE
MODULE dchunkmod
TYPE, PUBLIC :: dchunk
INTEGER :: asize
REAL(KIND=8), pointer :: xarr(:), yarr(:)
END TYPE dchunk
END MODULE
//C Example
struct dchunk{
int asize;
double* xarr, *yarr;
};
Then the pack-unpack subroutine is written as:
!FORTRAN EXAMPLE
SUBROUTINE dchunkpup(p, c)
USE pupmod
USE dchunkmod
IMPLICIT NONE
INTEGER :: p
TYPE(dchunk) :: c
pup(p, c%asize)
IF (fpup_isunpacking(p)) THEN !! if invocation is for unpacking
allocate(c%xarr(c%asize))
ALLOCATE(c%yarr(c%asize))
ENDIF
pup(p, c%xarr)
pup(p, c%yarr)
IF (fpup_isdeleting(p)) THEN !! if invocation is for packing
DEALLOCATE(c%xarr)
DEALLOCATE(c%yarr)
ENDIF
END SUBROUTINE
//C Example
void dchunkpup(pup_er p, struct dchunk c){
pup_int(p,c.asize);
if(pup_isUnpacking(p)){
c.xarr = (double *)malloc(sizeof(double)*c.asize);
c.yarr = (double *)malloc(sizeof(double)*c.asize);
}
pup_doubles(p,c.xarr,c.asize);
pup_doubles(p,c.yarr,c.asize);
if(pup_isPacking(p)){
free(c.xarr);
free(c.yarr);
}
}
One more function fpup_issizing is also available in module
pupmod that returns .TRUE. when the invocation is a sizing one.
In practice one almost never needs to use it.
Charm++ also provides higher-level PUP routines for C++ STL data structures and Fortran90 data types. The STL PUP routines will deduce the size of the structure automatically, so that the size of the data does not have to be passed in to the PUP routine. This facilitates writing PUP routines for large pre-existing codebases. To use it, simply include pup_stl.h in the user code. For modern Fortran with pointers and allocatable data types, AMPI provides a similarly automated PUP interface called apup. User code can include pupmod and then call apup() on any array (pointer or allocatable, multi-dimensional) of built-in types (character, short, int, long, real, double, complex, double complex, logical) and the runtime will deduce the size and shape of the array, including unassociated and NULL pointers. Here is the dchunk example from earlier, written to use the apup interface:
!FORTRAN EXAMPLE
SUBROUTINE dchunkpup(p, c)
USE pupmod
USE dchunkmod
IMPLICIT NONE
INTEGER :: p
TYPE(dchunk) :: c
!! no need for asize
!! no isunpacking allocation necessary
apup(p, c%xarr)
apup(p, c%yarr)
!! no isdeleting deallocation necessary
END SUBROUTINE
Calling MPI_ routines or accessing global variables that have been
privatized by use of tlsglobals or swapglobals from inside a user PUP
routine is currently not allowed in AMPI. Users can store MPI-related
information like communicator rank and size in data structures to be be
packed and unpacked before they are needed inside a PUP routine.
3.3.6. Extensions for Checkpointing¶
The pack-unpack subroutines written for migrations make sure that the current state of the program is correctly packed (serialized) so that it can be restarted on a different processor. Using the same subroutines, it is also possible to save the state of the program to disk, so that if the program were to crash abruptly, or if the allocated time for the program expires before completing execution, the program can be restarted from the previously checkpointed state. Thus, the pack-unpack subroutines act as the key facility for checkpointing in addition to their usual role for migration. Just as in load balancing, no application specific code is required when using Isomalloc: the AMPI runtime takes care of all the details involved in migrating data.
To perform a checkpoint in an AMPI program, all you have to do is make a
call to int AMPI_Migrate(MPI_Info hints) with an MPI_Info object
that specifies how you would like to checkpoint. Checkpointing can be
thought of as migrating AMPI ranks to storage. Users set the
checkpointing policy on an MPI_Info object’s "ampi_checkpoint"
key to one of the following values: "to_file=directory_name" or
"false". To perform checkpointing in memory a built-in MPI_Info
object called AMPI_INFO_CHKPT_IN_MEMORY is provided.
Checkpointing to file tells the runtime system to save checkpoints in a
given directory. (Typically, in an iterative program, the iteration
number, converted to a character string, can serve as a checkpoint
directory name.) This directory is created, and the entire state of the
program is checkpointed to this directory. One can restart the program
from the checkpointed state (using the same, more, or fewer physical
processors than were checkpointed with) by specifying
"+restart directory_name" on the command-line.
Checkpointing in memory allows applications to transparently tolerate failures online. The checkpointing scheme used here is a double in-memory checkpoint, in which virtual processors exchange checkpoints pairwise across nodes in each other’s memory such that if one node fails, that failed node’s AMPI ranks can be restarted by its buddy once the failure is detected by the runtime system. As long as no two buddy nodes fail in the same checkpointing interval, the system can restart online without intervention from the user (provided the job scheduler does not revoke its allocation). Any load imbalance resulting from the restart can then be managed by the runtime system. Use of this scheme is illustrated in the code snippet below.
// Main time-stepping loop
for (int iter=0; iter < max_iters; iter++) {
// Time step work ...
if (iter % chkpt_freq == 0)
AMPI_Migrate(AMPI_INFO_CHKPT_IN_MEMORY);
}
A value of "false" results in no checkpoint being done that step.
Note that AMPI_Migrate is a collective function, meaning every
virtual processor in the program needs to call this subroutine with the
same MPI_Info object. The checkpointing capabilities of AMPI are powered
by the Charm++ runtime system. For more information about
checkpoint/restart mechanisms please refer to the Charm++
manual: 2.3.12.
3.3.7. Extensions for Memory Efficiency¶
MPI functions usually require the user to preallocate the data buffers
needed before the functions being called. For unblocking communication
primitives, sometimes the user would like to do lazy memory allocation
until the data actually arrives, which gives the opportunities to write
more memory efficient programs. We provide a set of AMPI functions as an
extension to the standard MPI-2 one-sided calls, where we provide a
split phase MPI_Get called AMPI_Iget. AMPI_Iget preserves
the similar semantics as MPI_Get except that no user buffer is
provided to hold incoming data. AMPI_Iget_wait will block until the
requested data arrives and runtime system takes care to allocate space,
do appropriate unpacking based on data type, and return.
AMPI_Iget_free lets the runtime system free the resources being used
for this get request including the data buffer. Finally,
AMPI_Iget_data is the routine used to access the data.
int AMPI_Iget(MPI_Aint orgdisp, int orgcnt, MPI_Datatype orgtype, int rank,
MPI_Aint targdisp, int targcnt, MPI_Datatype targtype, MPI_Win win,
MPI_Request *request);
int AMPI_Iget_wait(MPI_Request *request, MPI_Status *status, MPI_Win win);
int AMPI_Iget_free(MPI_Request *request, MPI_Status *status, MPI_Win win);
int AMPI_Iget_data(void *data, MPI_Status status);
3.3.8. Extensions for Interoperability¶
Interoperability between different modules is essential for coding
coupled simulations. In this extension to AMPI, each MPI application
module runs within its own group of user-level threads distributed over
the physical parallel machine. In order to let AMPI know which ranks are
to be created, and in what order, a top level registration routine needs
to be written. A real-world example will make this clear. We have an MPI
code for fluids and another MPI code for solids, both with their main
programs, then we first transform each individual code to run correctly
under AMPI as standalone codes. Aside from the global and static
variable privatization transformations needed, this also involves making
the main program into a subroutine and naming it MPI_Main.
Thus now, we have two MPI_Mains, one for the fluids code and one
for the solids code. We now make these codes co-exist within the same
executable, by first renaming these MPI_Mains as Fluids_Main
and Solids_Main [5] writing a subroutine called MPI_Setup.
!FORTRAN EXAMPLE
SUBROUTINE MPI_Setup
USE ampi
CALL AMPI_Register_main(Solids_Main)
CALL AMPI_Register_main(Fluids_Main)
END SUBROUTINE
//C Example
void MPI_Setup(){
AMPI_Register_main(Solids_Main);
AMPI_Register_main(Fluids_Main);
}
This subroutine is called from the internal initialization routines of AMPI and tells AMPI how many numbers of distinct modules exist, and which orchestrator subroutines they execute.
The number of ranks to create for each module is specified on the
command line when an AMPI program is run. Appendix B explains how AMPI
programs are run, and how to specify the number of ranks (+vp
option). In the above case, suppose one wants to create 128 ranks of
Solids and 64 ranks of Fluids on 32 physical processors, one would
specify those with multiple +vp options on the command line as:
$ ./charmrun gen1.x +p 32 +vp 128 +vp 64
This will ensure that multiple modules representing different complete
applications can co-exist within the same executable. They can also
continue to communicate among their own ranks using the same AMPI
function calls to send and receive with communicator argument as
MPI_COMM_WORLD. But this would be completely useless if these
individual applications cannot communicate with each other, which is
essential for building efficient coupled codes. For this purpose, we
have extended the AMPI functionality to allow multiple
“COMM_WORLDs”; one for each application. These world
communicators form a “communicator universe” an array of communicators
aptly called MPI_COMM_UNIVERSE. This array of communicators is indexed
[1 … MPI_MAX_COMM]. In the current implementation,
MPI_MAX_COMM is 8, that is, maximum of 8 applications can co-exist
within the same executable.
The order of these COMM_WORLDs within MPI_COMM_UNIVERSE is
determined by the order in which individual applications are registered
in MPI_Setup.
Thus, in the above example, the communicator for the Solids module would
be MPI_COMM_UNIVERSE(1) and communicator for Fluids module would be
MPI_COMM_UNIVERSE(2).
Now any rank within one application can communicate with any rank in the other application using the familiar send or receive AMPI calls by specifying the appropriate communicator and the rank number within that communicator in the call. For example if a Solids rank number 36 wants to send data to rank number 47 within the Fluids module, it calls:
!FORTRAN EXAMPLE
INTEGER , PARAMETER :: Fluids_Comm = 2
CALL MPI_Send(InitialTime, 1, MPI_Double_Precision, tag,
47, MPI_Comm_Universe(Fluids_Comm), ierr)
//C Example
int Fluids_Comm = 2;
ierr = MPI_Send(InitialTime, 1, MPI_DOUBLE, tag,
47, MPI_Comm_Universe(Fluids_Comm));
The Fluids rank has to issue a corresponding receive call to receive this data:
!FORTRAN EXAMPLE
INTEGER , PARAMETER :: Solids_Comm = 1
CALL MPI_Recv(InitialTime, 1, MPI_Double_Precision, tag,
36, MPI_Comm_Universe(Solids_Comm), stat, ierr)
//C Example
int Solids_Comm = 1;
ierr = MPI_Recv(InitialTime, 1, MPI_DOUBLE, tag,
36, MPI_Comm_Universe(Solids_Comm), &stat);
3.3.9. Charm++ Interoperability¶
There is preliminary support for interoperating AMPI programs with Charm++
programs. This allows users to launch an AMPI program with an arbitrary number
of virtual processes in the same executable as a Charm++ program that contains
arbitrary collections of chares, with both AMPI ranks and chares being co-scheduled
by the runtime system. We also provide an entry method void injectMsg(int n, char buf[n])
for chares to communicate with AMPI ranks. An example program can be found in
examples/charm++/AMPI-interop.
3.3.10. Extensions for Sequential Re-run of a Parallel Node¶
In some scenarios, a sequential re-run of a parallel node is desired. One example is instruction-level accurate architecture simulations, in which case the user may wish to repeat the execution of a node in a parallel run in the sequential simulator. AMPI provides support for such needs by logging the change in the MPI environment on a certain processors. To activate the feature, build AMPI module with variable “AMPIMSGLOG” defined, like the following command in charm directory. (Linking with zlib “-lz” might be required with this, for generating compressed log file.)
$ ./build AMPI netlrts-linux-x86_64 -DAMPIMSGLOG
The feature is used in two phases: writing (logging) the environment and repeating the run. The first logging phase is invoked by a parallel run of the AMPI program with some additional command line options.
$ ./charmrun ./pgm +p4 +vp4 +msgLogWrite +msgLogRank 2 +msgLogFilename "msg2.log"
In the above example, a parallel run with 4 worker threads and 4 AMPI ranks will be executed, and the changes in the MPI environment of worker thread 2 (also rank 2, starting from 0) will get logged into diskfile “msg2.log”.
Unlike the first run, the re-run is a sequential program, so it is not invoked by charmrun (and omitting charmrun options like +p4 and +vp4), and additional command line options are required as well.
$ ./pgm +msgLogRead +msgLogRank 2 +msgLogFilename "msg2.log"
3.3.11. User Defined Initial Mapping¶
You can define the initial mapping of virtual processors (vp) to physical processors (p) as a runtime option. You can choose from predefined initial mappings or define your own mappings. The following predefined mappings are available:
- Round Robin
This mapping scheme maps virtual processor to physical processor in round-robin fashion, i.e. if there are 8 virtual processors and 2 physical processors then virtual processors indexed 0,2,4,6 will be mapped to physical processor 0 and virtual processors indexed 1,3,5,7 will be mapped to physical processor 1.
$ ./charmrun ./hello +p2 +vp8 +mapping RR_MAP
- Block Mapping
This mapping scheme maps virtual processors to physical processor in ranks, i.e. if there are 8 virtual processors and 2 physical processors then virtual processors indexed 0,1,2,3 will be mapped to physical processor 0 and virtual processors indexed 4,5,6,7 will be mapped to physical processor 1.
$ ./charmrun ./hello +p2 +vp8 +mapping BLOCK_MAP
- Proportional Mapping
This scheme takes the processing capability of physical processors into account for mapping virtual processors to physical processors, i.e. if there are 2 processors running at different frequencies, then the number of virtual processors mapped to processors will be in proportion to their processing power. To make the load balancing framework aware of the heterogeneity of the system, the flag +LBTestPESpeed should also be used.
$ ./charmrun ./hello +p2 +vp8 +mapping PROP_MAP $ ./charmrun ./hello +p2 +vp8 +mapping PROP_MAP +balancer GreedyLB +LBTestPESpeed
If you want to define your own mapping scheme, please contact us for assistance.
3.3.12. Performance Visualization¶
AMPI users can take advantage of Charm++’s tracing framework and associated performance visualization tool, Projections. Projections provides a number of different views of performance data that help users diagnose performance issues. Along with the traditional Timeline view, Projections also offers visualizations of load imbalance and communication-related data.
In order to generate tracing logs from an application to view in
Projections, link with ampicc -tracemode projections.
AMPI defines the following extensions for tracing support:
AMPI_Trace_begin AMPI_Trace_end
When using the Timeline view in Projections, AMPI users can visualize what each VP on each processor is doing (what MPI method it is running or blocked in) by clicking the View tab and then selecting Show Nested Bracketed User Events from the drop down menu. See the Projections manual for information on performance analysis and visualization.
AMPI users can also use any tracing libraries or tools that rely on MPI’s PMPI profiling interface, though such tools may not be aware of AMPI process virtualization.
3.3.13. Compiling AMPI Programs¶
AMPI provides a cross-platform compile-and-link script called ampicc
to compile C, C++, and Fortran AMPI programs. This script resides in the
bin subdirectory in the Charm++ installation directory. The main
purpose of this script is to deal with the differences of various
compiler names and command-line options across various machines on which
AMPI runs. It is recommended that the AMPI compiler scripts be used to
compile and link AMPI programs. One major advantage of using these is
that one does not have to specify which libraries are to be linked for
ensuring that C++ and Fortran90 codes are linked together correctly.
Appropriate libraries required for linking such modules together are
known to ampicc for various machines.
In spite of the platform-neutral syntax of ampicc, one may have to specify some platform-specific options for compiling and building AMPI codes. Fortunately, if ampicc does not recognize any particular options on its command line, it promptly passes it to all the individual compilers and linkers it invokes to compile the program. See the appendix for more details on building and running AMPI programs.
3.3.14. AMPI Example Applications¶
git clone ssh://charm.cs.illinois.edu:9418/benchmarks/ampi-benchmarks.Most benchmarks can be compiled with the provided top-level Makefile:
$ git clone ssh://charm.cs.illinois.edu:9418/benchmarks/ampi-benchmarks
$ cd ampi-benchmarks
$ make -f Makefile.ampi
3.3.14.1. Mantevo project v3.0¶
Set of mini-apps from the Mantevo project. Download at https://mantevo.org/download/.
3.3.14.1.1. MiniFE¶
- Mantevo mini-app for unstructured implicit Finite Element computations.
- No changes necessary to source to run on AMPI. Modify file
makefile.ampiand change variableAMPIDIRto point to your Charm++ directory, executemake -f makefile.ampito build the program. - Refer to the
READMEfile on how to run the program. For example:./charmrun +p4 ./miniFE.x nx=30 ny=30 nz=30 +vp32
3.3.14.1.2. MiniMD v2.0¶
- Mantevo mini-app for particle interaction in a Lennard-Jones system, as in the LAMMPS MD code.
- No changes necessary to source code. Modify file
Makefile.ampiand change variableAMPIDIRto point to your Charm++ directory, executemake ampito build the program. - Refer to the
READMEfile on how to run the program. For example:./charmrun +p4 ./miniMD_ampi +vp32
3.3.14.1.3. CoMD v1.1¶
- Mantevo mini-app for molecular dynamics codes: https://github.com/exmatex/CoMD
- To AMPI-ize it, we had to remove calls to not thread-safe
getopt(). Support for dynamic load balancing has been added in the main loop and the command line options. It will run on all platforms. - Just update the Makefile to point to AMPI compilers and run with the provided run scripts.
3.3.14.1.4. MiniXYCE v1.0¶
- Mantevo mini-app for discrete analog circuit simulation, version 1.0, with serial, MPI, OpenMP, and MPI+OpenMP versions.
- No changes besides Makefile necessary to run with virtualization. To
build, do
cp common/generate_info_header miniXyce_ref/., modify the CC path inminiXyce_ref/and runmake. Run scripts are intest/. - Example run command:
./charmrun +p3 ./miniXyce.x +vp3 -circuit ../tests/cir1.net -t_start 1e-6 -pf params.txt
3.3.14.1.5. HPCCG v1.0¶
- Mantevo mini-app for sparse iterative solves using the Conjugate Gradient method for a problem similar to that of MiniFE.
- No changes necessary except to set compilers in
Makefileto the AMPI compilers. - Run with a command such as:
./charmrun +p2 ./test_HPCCG 20 30 10 +vp16
3.3.14.1.6. MiniAMR v1.0¶
- miniAMR applies a stencil calculation on a unit cube computational domain, which is refined over time.
- No changes if using swap-globals. Explicitly extern global variables if using TLS.
3.3.14.1.7. Not yet AMPI-zed (reason):¶
MiniAero v1.0 (build issues), MiniGhost v1.0.1 (globals), MiniSMAC2D v2.0 (globals), TeaLeaf v1.0 (globals), CloverLeaf v1.1 (globals), CloverLeaf3D v1.0 (globals).
3.3.14.2. LLNL ASC Proxy Apps¶
3.3.14.2.1. LULESH v2.0¶
- LLNL Unstructured Lagrangian-Eulerian Shock Hydrodynamics proxy app: https://codesign.llnl.gov/lulesh.php
- Charm++, MPI, MPI+OpenMP, Liszt, Loci, Chapel versions all exist for comparison.
- Manually privatized version of LULESH 2.0, plus a version with PUP
routines in subdirectory
pup_lulesh202/.
3.3.14.2.2. AMG 2013¶
- LLNL ASC proxy app: Algebraic Multi-Grid solver for linear systems arising from unstructured meshes: https://codesign.llnl.gov/amg2013.php
- AMG is based on HYPRE, both from LLNL. The only change necessary to get AMG running on AMPI with virtualization is to remove calls to HYPRE’s timing interface, which is not thread-safe.
- To build, point the CC variable in Makefile.include to your AMPI CC
wrapper script and
make. Executable istest/amg2013.
3.3.14.2.3. Lassen v1.0¶
- LLNL ASC mini-app for wave-tracking applications with dynamic load imbalance. Reference versions are serial, MPI, Charm++, and MPI/Charm++ interop: https://codesign.llnl.gov/lassen.php
- No changes necessary to enable AMPI virtualization. Requires some
C++11 support. Set
AMPIDIRin Makefile andmake. Run with:./charmrun +p4 ./lassen_mpi +vp8 default 2 2 2 50 50 50
3.3.14.2.4. Kripke v1.1¶
LLNL ASC proxy app for ARDRA, a full Sn deterministic particle transport application: https://codesign.llnl.gov/kripke.php
Charm++, MPI, MPI+OpenMP, MPI+RAJA, MPI+CUDA, MPI+OCCA versions exist for comparison.
Kripke requires no changes between MPI and AMPI since it has no global/static variables. It uses cmake so edit the cmake toolchain files in
cmake/toolchain/to point to the AMPI compilers, and build in a build directory:$ mkdir build; cd build; $ cmake .. -DCMAKE_TOOLCHAIN_FILE=../cmake/Toolchain/linux-gcc-ampi.cmake -DENABLE_OPENMP=OFF $ make
Run with:
$ ./charmrun +p8 ./src/tools/kripke +vp8 --zones 64,64,64 --procs 2,2,2 --nest ZDG
3.3.14.2.5. MCB v1.0.3 (2013)¶
LLNL ASC proxy app for Monte Carlo particle transport codes: https://codesign.llnl.gov/mcb.php
MPI+OpenMP reference version.
Run with:
$ OMP_NUM_THREADS=1 ./charmrun +p4 ./../src/MCBenchmark.exe --weakScaling --distributedSource --nCores=1 --numParticles=20000 --multiSigma --nThreadCore=1 +vp16
3.3.14.2.6. Not yet AMPI-zed (reason)¶
: UMT 2013 (global variables).
3.3.14.3. Other Applications¶
3.3.14.3.1. MILC 7.0¶
- MILC is a code to study quantum chromodynamics (QCD) physics. http://www.nersc.gov/users/computational-systems/cori/nersc-8-procurement/trinity-nersc-8-rfp/nersc-8-trinity-benchmarks/milc/
- Moved
MPI_Init_threadcall tomain(), added__threadto all global/static variable declarations. Runs on AMPI with virtualization when using -tlsglobals. - Build: edit
ks_imp_ds/Makefileto use AMPI compiler wrappers, runmake su3_rmdinks_imp_ds/ - Run with:
./su3_rmd +vp8 ../benchmark_n8/single_node/n8_single.in
3.3.14.3.2. SNAP v1.01 (C version)¶
- LANL proxy app for PARTISN, an Sn deterministic particle transport application: https://github.com/losalamos/SNAP
- SNAP is an update to Sweep3D. It simulates the same thing as Kripke, but with a different decomposition and slight algorithmic differences. It uses a 1- or 2-dimensional decomposition and the KBA algorithm to perform parallel sweeps over the 3-dimensional problem space. It contains all of the memory, computation, and network performance characteristics of a real particle transport code.
- Original SNAP code is Fortran90-MPI-OpenMP, but this is a C-MPI-OpenMP version of it provided along with the original version. The Fortran90 version will require global variable privatization, while the C version works out of the box on all platforms.
- Edit the Makefile for AMPI compiler paths and run with:
./charmrun +p4 ./snap +vp4 --fi center_src/fin01 --fo center_src/fout01
3.3.14.3.3. Sweep3D¶
- Sweep3D is a particle transport program that analyzes the flux of particles along a space. It solves a three-dimensional particle transport problem.
- This mini-app has been deprecated, and replaced at LANL by SNAP (above).
- Build/Run Instructions:
- Modify the
makefileand change variable CHARMC to point to your Charm++ compiler command, executemake mpito build the program. - Modify file
inputto set the different parameters. Refer to fileREADMEon how to change those parameters. Run with:./charmrun ./sweep3d.mpi +p8 +vp16
- Modify the
3.3.14.3.4. PENNANT v0.8¶
- Unstructured mesh Rad-Hydro mini-app for a full application at LANL called FLAG. https://github.com/losalamos/PENNANT
- Written in C++, only global/static variables that need to be privatized are mype and numpe. Done manually.
- Legion, Regent, MPI, MPI+OpenMP, MPI+CUDA versions of PENNANT exist for comparison.
- For PENNANT-v0.8, point CC in Makefile to AMPICC and just ’make’. Run
with the provided input files, such as:
./charmrun +p2 ./build/pennant +vp8 test/noh/noh.pnt
3.3.14.4. Benchmarks¶
3.3.14.4.1. Jacobi-2D (Fortran)¶
- Jacobi-2D with 1D decomposition. Problem size and number of iterations are defined in the source code. Manually privatized.
3.3.14.4.2. Jacobi-3D (C)¶
- Jacobi-3D with 3D decomposition. Manually privatized. Includes multiple versions: Isomalloc, PUP, FT, LB, Isend/Irecv, Iput/Iget.
3.3.14.4.3. NAS Parallel Benchmarks (NPB 3.3)¶
- A collection of kernels used in different scientific applications. They are mainly implementations of various linear algebra methods. http://www.nas.nasa.gov/Resources/Software/npb.html
- Build/Run Instructions:
- Modify file
config/make.defto make variableCHAMRDIRpoint to the right Charm++ directory. - Use
make <benchmark> NPROCS=<P> CLASS=<C>to build a particular benchmark. The values for<benchmark>are (bt, cg, dt, ep, ft, is, lu, mg, sp),<P>is the number of ranks and<C>is the class or the problem size (to be chosen from A,B,C,D or E). Some benchmarks may have restrictions on values of<P>and<C>. For instance, to make CG benchmark with 256 ranks and class C, we will use the following command:make cg NPROCS=256 - The resulting executable file will be generated in the respective
directory for the benchmark. In the previous example, a file
cg.256.C will appear in the CG and
bin/directories. To run the particular benchmark, you must follow the standard procedure of running AMPI programs:./charmrun ./cg.C.256 +p64 +vp256 ++nodelist nodelist +isomalloc_sync
- Modify file
3.3.14.4.4. NAS PB Multi-Zone Version (NPB-MZ 3.3)¶
- A multi-zone version of BT, SP and LU NPB benchmarks. The multi-zone intentionally divides the space unevenly among ranks and causes load imbalance. The original goal of multi-zone versions was to offer an test case for hybrid MPI+OpenMP programming, where the load imbalance can be dealt with by increasing the number of threads in those ranks with more computation. http://www.nas.nasa.gov/Resources/Software/npb.html
- The BT-MZ program shows the heaviest load imbalance.
- Build/Run Instructions:
- Modify file
config/make.defto make variableCHAMRDIRpoint to the right Charm++ build. - Use the format
make <benchmark> NPROCS=<P> CLASS=<C>to build a particular benchmark. The values for<benchmark>are (bt-mz, lu-mz, sp-mz),<P>is the number of ranks and<C>is the class or the problem size (to be chosen from A,B,C,D or E). Some benchmarks may have restrictions on values of<P>and<C>. For instance, to make the BT-MZ benchmark with 256 ranks and class C, you can use the following command:make bt-mz NPROCS=256 CLASS=C - The resulting executable file will be generated in the bin/
directory. In the previous example, a file bt-mz.256.C will be
created in the
bindirectory. To run the particular benchmark, you must follow the standard procedure of running AMPI programs:./charmrun ./bt-mz.C.256 +p64 +vp256 ++nodelist nodelist +isomalloc_sync
- Modify file
3.3.14.4.5. HPCG v3.0¶
- High Performance Conjugate Gradient benchmark, version 3.0. Companion metric to Linpack, with many vendor-optimized implementations available: http://hpcg-benchmark.org/
- No AMPI-ization needed. To build, modify
setup/Make.AMPIfor compiler paths, domkdir build && cd build && configure ../setup/Make.AMPI && make. To run, do./charmrun +p16 ./bin/xhpcg +vp64
3.3.14.4.6. Intel Parallel Research Kernels (PRK) v2.16¶
- A variety of kernels (Branch, DGEMM, Nstream, Random, Reduce, Sparse, Stencil, Synch_global, Synch_p2p, and Transpose) implemented for a variety of runtimes (SERIAL, OpenMP, MPI-1, MPI-RMA, MPI-SHM, MPI+OpenMP, SHMEM, FG_MPI, UPC, Grappa, Charm++, and AMPI). https://github.com/ParRes/Kernels
- For AMPI tests, set
CHARMTOPand run:make allampi. There are run scripts included.
3.3.14.4.7. OSU Microbenchmarks¶
MPI collectives performance testing suite. https://charm.cs.illinois.edu/gerrit/#/admin/projects/benchmarks/osu-collectives-benchmarking
- Build with:
./configure CC=~/charm/bin/ampicc && make
3.3.14.5. Third Party Open Source Libraries¶
3.3.14.5.1. HYPRE-2.11.1¶
High Performance Preconditioners and solvers library from LLNL. https://computation.llnl.gov/project/linear_solvers/software.php
Hypre-2.11.1 builds on top of AMPI using the configure command:
$ ./configure --with-MPI \ CC=~/charm/bin/ampicc \ CXX=~/charm/bin/ampicxx \ F77=~/charm/bin/ampif77 \ --with-MPI-include=~/charm/include \ --with-MPI-lib-dirs=~/charm/lib \ --with-MPI-libs=mpi --without-timing --without-print-errors $ make -j8
All HYPRE tests and examples pass tests with virtualization, migration, etc. except for those that use Hypre’s timing interface, which uses a global variable internally. So just remove those calls and do not define
HYPRE_TIMINGwhen compiling a code that uses Hypre. In the examples directory, you’ll have to set the compilers to your AMPI compilers explicitly too. In the test directory, you’ll have to edit the Makefile to 1) Remove-DHYPRE_TIMINGfrom bothCDEFSandCXXDEFS, 2) Remove both${MPILIBS}and${MPIFLAGS}fromMPILIBFLAGS, and 3) Remove${LIBS}fromLIBFLAGS. Then runmake.To run the
new_ijtest, run:./charmrun +p64 ./new_ij -n 128 128 128 -P 4 4 4 -intertype 6 -tol 1e-8 -CF 0 -solver 61 -agg_nl 1 27pt -Pmx 6 -ns 4 -mu 1 -hmis -rlx 13 +vp64
3.3.14.5.2. MFEM-3.2¶
- MFEM is a scalable library for Finite Element Methods developed at LLNL. http://mfem.org/
- MFEM-3.2 builds on top of AMPI (and METIS-4.0.3 and HYPRE-2.11.1). Download MFEM, HYPRE, and METIS. Untar all 3 in the same top-level directory.
- Build HYPRE-2.11.1 as described above.
- Build METIS-4.0.3 by doing
cd metis-4.0.3/ && make - Build MFEM-3.2 serial first by doing
make serial - Build MFEM-3.2 parallel by doing:
- First, comment out
#define HYPRE_TIMINGinmfem/linalg/hypre.hpp. Also, you must add a#define hypre_clearTiming()at the top oflinalg/hypre.cpp, because Hypre-2.11.1 has a bug where it doesn’t provide a definition of this function if you don’t defineHYPRE_TIMING. make parallel MFEM_USE_MPI=YES MPICXX=~/charm/bin/ampicxx HYPRE_DIR=~/hypre-2.11.1/src/hypre METIS_DIR=~/metis-4.0.3
- First, comment out
- To run an example, do
./charmrun +p4 ./ex15p -m ../data/amr-quad.mesh +vp16. You may want to add the runtime options-no-visand-no-visitto speed things up. - All example programs and miniapps pass with virtualization, and migration if added.
3.3.14.5.3. XBraid-1.1¶
- XBraid is a scalable library for parallel time integration using MultiGrid, developed at LLNL. https://computation.llnl.gov/project/parallel-time-integration/software.php
- XBraid-1.1 builds on top of AMPI (and its examples/drivers build on top of MFEM-3.2, HYPRE-2.11.1, and METIS-4.0.3 or METIS-5.1.0).
- To build XBraid, modify the variables CC, MPICC, and MPICXX in
makefile.inc to point to your AMPI compilers, then do
make. - To build XBraid’s examples/ and drivers/ modify the paths to MFEM and
HYPRE in their Makefiles and
make. - To run an example, do
./charmrun +p2 ./ex-02 -pgrid 1 1 8 -ml 15 -nt 128 -nx 33 33 -mi 100 +vp8 ++local. - To run a driver, do
./charmrun +p4 ./drive-03 -pgrid 2 2 2 2 -nl 32 32 32 -nt 16 -ml 15 +vp16 ++local
3.3.14.6. Other AMPI codes¶
- FLASH
- BRAMS (Weather prediction model)
- CGPOP
- Fractography3D (Crack Propagation)
- JetAlloc
- PlasComCM (XPACC)
- PlasCom2 (XPACC)
- Harm3D
3.4. Installing AMPI¶
AMPI is included in the source distribution of Charm++. To get the latest sources from PPL, visit: http://charm.cs.illinois.edu/software
and follow the download links. Then build Charm++ and AMPI from source.
The build script for Charm++ is called build. The syntax for this
script is:
$ build <target> <version> <opts>
For building AMPI (which also includes building Charm++ and other
libraries needed by AMPI), specify <target> to be AMPI. And
<opts> are command line options passed to the charmc compile
script. Common compile time options such as
-g, -O, -Ipath, -Lpath, -llib are accepted.
To build a debugging version of AMPI, use the option: -g. To build a
production version of AMPI, use the option: -with-production.
<version> depends on the machine, operating system, and the
underlying communication library one wants to use for running AMPI
programs. See the charm/README file for details on picking the proper
version. Here is an example of how to build a debug version of AMPI in a
linux and ethernet environment:
$ ./build AMPI netlrts-linux-x86_64 -g
And the following is an example of how to build a production version of AMPI on a Cray XC system, with MPI-level error checking in AMPI turned off:
$ ./build AMPI gni-crayxc --with-production --disable-ampi-error-checking
AMPI can also be built with support for shared memory on any communication layer by adding “smp” as an option after the build target. For example, on an Infiniband Linux cluster:
$ ./build AMPI verbs-linux-x86_64 smp --with-production
AMPI ranks are implemented as user-level threads with a stack size default of 1MB. If the default is not correct for your program, you can specify a different default stack size (in bytes) at build time. The following build command illustrates this for an Intel Omni-Path system:
$ ./build AMPI ofi-linux-x86_64 --with-production -DTCHARM_STACKSIZE_DEFAULT=16777216
The same can be done for AMPI’s RDMA messaging threshold using
AMPI_RDMA_THRESHOLD_DEFAULT and, for messages sent within the same
address space (ranks on the same worker thread or ranks on different
worker threads in the same process in SMP builds), using
AMPI_SMP_RDMA_THRESHOLD_DEFAULT. Contiguous messages with sizes
larger than the threshold are sent via RDMA on communication layers that
support this capability. You can also set the environment variables
AMPI_RDMA_THRESHOLD and AMPI_SMP_RDMA_THRESHOLD before running a
job to override the default specified at build time.
3.5. Building and Running AMPI Programs¶
3.5.1. Building AMPI Programs¶
AMPI provides a compiler called ampicc in your charm/bin/ directory. You can use this compiler to build your AMPI program the same way as other compilers like cc. All the command line flags that you would use for other compilers can be used with the AMPI compilers the same way. For example:
$ ampicc -c pgm.c -O3
$ ampif90 -c pgm.f90 -O0 -g
$ ampicc -o pgm pgm.o -lm -O3
To use Isomalloc for transparently migrating user heap data, link with -memory isomalloc. To use a Charm++ load balancer, link a strategy or a suite of strategies in with -module <LB>. For example:
$ ampicc pgm.c -o pgm -O3 -memory isomalloc -module CommonLBs
3.5.2. Running AMPI programs¶
AMPI offers two options to execute an AMPI program, charmrun and
ampirun.
3.5.2.1. Running with charmrun¶
The Charm++ distribution contains a script called charmrun that
makes the job of running AMPI programs portable and easier across all
parallel machines supported by Charm++. charmrun is copied to a
directory where an AMPI program is built using ampicc. It takes a
command line parameter specifying number of processors, and the name of
the program followed by AMPI options (such as number of ranks to create,
and the stack size of every user-level thread) and the program
arguments. A typical invocation of an AMPI program pgm with
charmrun is:
$ ./charmrun +p16 ./pgm +vp64
Here, the AMPI program pgm is run on 16 physical processors with 64
total virtual ranks (which will be mapped 4 per processor initially).
To run with load balancing, specify a load balancing strategy. If
Address Space Layout Randomization is enabled on your target system, you
may need to add the flag +isomalloc_sync when running with
migration. You can also specify the size of user-level thread’s stack
using the +tcharm_stacksize option, which can be used to decrease
the size of the stack that must be migrated, as in the following
example:
$ ./charmrun +p16 ./pgm +vp128 +tcharm_stacksize 32K +balancer RefineLB
3.5.2.2. Running with ampirun¶
For compliance with the MPI standard and simpler execution, AMPI ships
with the ampirun script that is similar to mpirun provided by
other MPI runtimes. As with charmrun, ampirun is copied
automatically to the program directory when compiling an application
with ampicc.
The basic usage of ampirun is as follows:
$ ./ampirun -np 16 --host h1,h2,h3,h4 ./pgm
This command will create 16 (non-virtualized) ranks and distribute them on the hosts h1-h4.
When using the -vr option, AMPI will create the number of ranks
specified by the -np parameter as virtual ranks, and will create
only one process per host:
$ ./ampirun -np 16 --host h1,h2,h3,h4 -vr ./pgm
Other options (such as the load balancing strategy), can be specified in the same way as for charmrun:
$ ./ampirun -np 16 ./pgm +balancer RefineLB
3.5.2.3. Other options¶
Note that for AMPI programs compiled with gfortran, users may need to set the following environment variable to see program output to stdout:
$ export GFORTRAN_UNBUFFERED_ALL=1
| [1] | Currently, AMPI supports the MPI-2.2 standard, and the MPI-3.1 standard is under active development, though we already support non-blocking and neighborhood collectives among other MPI-3.1 features. |
| [2] | http://www-unix.mcs.anl.gov/romio/ |
| [3] | http://www.eclipse.org/photran |
| [4] | http://rosecompiler.org/ |
| [5] | Currently, we assume that the interface code, which does mapping and interpolation among the boundary values of Fluids and Solids domain, is integrated with one of Fluids and Solids. |
| [PiP2018] | Atsushi Hori, Min Si, Balazs Gerofi, Masamichi Takagi, Jai Dayal, Pavan Balaji, and Yutaka Ishikawa. 2018. Process-in-process: techniques for practical address-space sharing. In Proceedings of the 27th International Symposium on High-Performance Parallel and Distributed Computing (HPDC ‘18). ACM, New York, NY, USA, 131-143. DOI: https://doi.org/10.1145/3208040.3208045 |